No Database? No Problem: Using discord with Simple Family Structures
Mason Garrison
Source:vignettes/links.Rmd
links.RmdIntroduction
The {discord} package was originally developed for use with the National Longitudinal Survey of Youth (NLSY), but its functionality extends far beyond that. When paired with its sister package {BGmisc}, discord can be applied to any dataset containing basic family structure information, allowing researchers to explore genetic and environmental influences without requiring pre-constructed kinship links.
This vignette demonstrates how to:
- Construct kinship links from simple family data (e.g., individual ID, mother ID, father ID).
- Simulate phenotyipic data based on known genetic and environmental structures.
- Fit a discordant-kinship regression model using the simulated data.
We use tools from {BGmisc} and a toy dataset to illustrate the workflow.
Loading Packages and Data
We begin by loading the required packages and a built-in dataset from {BGmisc}.
We rename the family ID column to avoid naming conflicts and generate a pedigree-encoded data frame.
df_potter <- potter
names(df_potter)[names(df_potter) == "famID"] <- "oldfam"
df_potter <- ped2fam(df_potter,
famID = "famID",
personID = "personID"
)We also verify and repair sex coding to ensure compatibility with downstream pedigree operations.
df_potter <- checkSex(df_potter,
code_male = 1,
code_female = 0,
verbose = FALSE, repair = TRUE
)
ggpedigree(potter, config = list(
label_method = "geom_text",
label_nudge_y = .25
)) +
labs(title = "Pedigree Plot of the Potter Dataset") +
theme(legend.position = "bottom")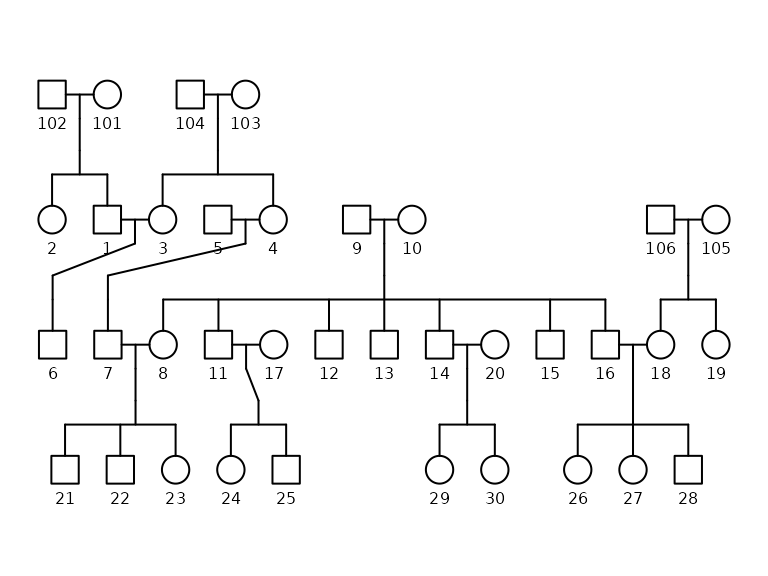
Pedigree plot of the Potter dataset
The pedigree plot provides a visual representation of the kinship structure in the dataset. Each node represents an individual, and the edges indicate familial relationships.
Constructing Kinship Links
To extract the necessary kinship information, we need to compute two matrices: the additive genetic relatedness matrix and the shared environment matrix. These matrices are derived from the pedigree data and are essential for understanding the genetic and environmental relationships among individuals.
Using {BGmisc}, we compute:
The additive genetic relatedness matrix (add).
The shared environment matrix (cn), indicating whether kin were raised together (1) or apart (0).
The ped2add() function computes the additive genetic
relatedness matrix, which quantifies the genetic similarity between
individuals based on their pedigree information. The
ped2cn() function computes the shared environment matrix,
which indicates whether individuals were raised in the same environment
(1) or different environments (0).
The resulting matrices are symmetric, with diagonal elements representing self-relatedness (1.0). The off-diagonal elements represent the relatedness between pairs of individuals, with values ranging from 0 (no relatedness) to 0.5 (full siblings) to 1 (themselves).
We convert the component matrices into a long-form data frame of kin
pairs using com2links(). Self-pairs and duplicate entries
are removed.
df_links <- com2links(
writetodisk = FALSE,
ad_ped_matrix = add,
cn_ped_matrix = cn,
drop_upper_triangular = TRUE
) %>%
filter(ID1 != ID2)
df_links %>%
slice(1:10) %>%
knitr::kable()| ID1 | ID2 | addRel | cnuRel |
|---|---|---|---|
| 1 | 2 | 0.500 | 1 |
| 3 | 4 | 0.500 | 1 |
| 1 | 6 | 0.500 | 0 |
| 2 | 6 | 0.250 | 0 |
| 3 | 6 | 0.500 | 0 |
| 4 | 6 | 0.250 | 0 |
| 3 | 7 | 0.250 | 0 |
| 4 | 7 | 0.500 | 0 |
| 5 | 7 | 0.500 | 0 |
| 6 | 7 | 0.125 | 0 |
As you can see, the df_links data frame contains pairs
of individuals (ID1 and ID2) along with their additive genetic
relatedness (addRel) and shared environment status
(cnuRel). These data are in wide format, with each row
representing a unique pair of individuals.
Further, we can tally the number of pairs by relatedness and shared environment to understand the composition of the dataset.
df_links %>%
group_by(addRel, cnuRel) %>%
tally()
#> # A tibble: 5 × 3
#> # Groups: addRel [4]
#> addRel cnuRel n
#> <dbl> <dbl> <int>
#> 1 0.0625 0 3
#> 2 0.125 0 47
#> 3 0.25 0 104
#> 4 0.5 0 50
#> 5 0.5 1 32As you can see, the dataset contains a variety of kinship pairs, including full siblings, parent-child, aunt-nephew, cousins, and unrelated individuals, with varying degrees of shared environment.
For this demonstration, we will focus on cousins. Cousins share some genetic relatedness but typically do not share the same environment. (In contrast, full siblings share both genetic relatedness and environment. Half-siblings share genetic relatedness but sometimes do not share the same environment, making them less ideal for this demonstration.)
We then extract two subsets:
Full siblings: additive relatedness = 0.5 and shared environment = 1
Cousins: additive relatedness = 0.125 and shared environment = 0
df_siblings <- df_links %>%
filter(addRel == .5) %>% # only full siblings %>%
filter(cnuRel == 1) # only kin raised in the same home
df_cousin <- df_links %>%
filter(addRel == .125) %>% # only cousins %>%
filter(cnuRel == 0) # only kin raised in separate homesNow for the rest of the vignette, we will use the cousin subset
(df_cousin) to illustrate the process of simulating
phenotypic data and fitting a discordant-kinship regression model.
However, given the small sample size of cousins in this dataset, we will
simulate four datasets worth of cousins and combine them to increase the
sample size.
Simulate Phenotypic Data
To simulate phenotypic data, we need to create a data frame that
includes the kinship information and the outcome variables. We will
simulate two outcome variables (y1 and y2) for each kin pair in the
dataset. The kinsim() function from {discord} is used to
generate the simulated data based on the specified variance structure.
For convenience, we will generate data for all the cousins in
df_links. However, we could also generate data for the full
siblings or any other kinship pairs.
set.seed(1234)
syn_df <- discord::kinsim(
mu_all = c(2, 2),
cov_a = .4,
cov_e = .4,
c_vector = df_cousin$cnuRel,
r_vector = df_cousin$addRel
) %>%
select(-c(
A1_1, A1_2, A2_1, A2_2,
C1_1, C1_2, C2_1, C2_2,
E1_1, E1_2, E2_1, E2_2,
r
))The simulated data reflect a known variance structure: additive genetic covariance = .4, genetic relatedness of 0.125, no shared environment, and residual (unique environment) variance = 0.4. Latent component scores are dropped from the final dataset, but they can be useful for debugging and understanding the underlying structure of the data.
We bind the simulated outcome data to the links data to prepare it for modeling.
data_demo <- cbind(df_cousin, syn_df) %>%
arrange(ID1, ID2)
summary(data_demo)
#> ID1 ID2 addRel cnuRel
#> Min. : 3.0 Min. : 7.0 Min. :0.125 Min. :0
#> 1st Qu.: 759.2 1st Qu.: 781.2 1st Qu.:0.125 1st Qu.:0
#> Median :1515.5 Median :1555.5 Median :0.125 Median :0
#> Mean :1521.6 Mean :1536.6 Mean :0.125 Mean :0
#> 3rd Qu.:2271.8 3rd Qu.:2329.8 3rd Qu.:0.125 3rd Qu.:0
#> Max. :3028.0 Max. :3104.0 Max. :0.125 Max. :0
#> y1_1 y1_2 y2_1 y2_2
#> Min. :-3.3672 Min. :-1.5255 Min. :-2.959 Min. :-2.3276
#> 1st Qu.: 0.6044 1st Qu.: 0.8779 1st Qu.: 0.787 1st Qu.: 0.9394
#> Median : 1.7633 Median : 2.1013 Median : 1.847 Median : 2.0066
#> Mean : 1.7469 Mean : 2.0855 Mean : 2.039 Mean : 2.1207
#> 3rd Qu.: 2.9137 3rd Qu.: 3.1359 3rd Qu.: 3.208 3rd Qu.: 3.3210
#> Max. : 6.0358 Max. : 6.1543 Max. : 6.783 Max. : 6.7877
#> id
#> Min. : 1.00
#> 1st Qu.: 47.75
#> Median : 94.50
#> Mean : 94.50
#> 3rd Qu.:141.25
#> Max. :188.00| ID1 | ID2 | addRel | cnuRel | y1_1 | y1_2 | y2_1 | y2_2 | id |
|---|---|---|---|---|---|---|---|---|
| 3 | 21 | 0.125 | 0 | 0.6984681 | -0.9463635 | 0.9203754 | 0.8795634 | 2 |
| 3 | 22 | 0.125 | 0 | 3.4082317 | 0.1747639 | 4.5457972 | 0.2776425 | 3 |
| 3 | 23 | 0.125 | 0 | -1.9437220 | -1.4571830 | 2.8360088 | 2.4356576 | 4 |
| 6 | 7 | 0.125 | 0 | 3.0188293 | 5.1542516 | 0.7041064 | 0.9333481 | 1 |
| 21 | 24 | 0.125 | 0 | 0.5056531 | 0.0700135 | 1.1275003 | 4.2852528 | 5 |
The data_demo data frame now contains the kinship
information along with the simulated outcome variables y1 and y2. Each
row represents a pair of cousins, and the columns include the IDs of the
individuals, their relatedness, and the simulated phenotypic data.
Fitting a Discordant-Kinship Regression Model
We then use discord_regression() to fit a
discordant-kinship model, predicting y1 from y2. Based on the structure
of the data, we expect that there will be a significant association
between the two outcome variables, as there is a known overlapping
non-shared environment covariance.
The model is fit using the discord_regression()
function, which takes the following arguments:
model_output <- discord_regression(
data = data_demo,
outcome = "y1",
predictors = "y2",
id = "id",
sex = NULL,
race = NULL,
pair_identifiers = c("_1", "_2")
)
summary(model_output)
#>
#> Call:
#> stats::lm(formula = y1_diff ~ y1_mean + y2_diff + y2_mean, data = preppedData)
#>
#> Residuals:
#> Min 1Q Median 3Q Max
#> -1.8511 -0.8476 -0.1705 0.6317 3.6083
#>
#> Coefficients:
#> Estimate Std. Error t value Pr(>|t|)
#> (Intercept) 1.38903 0.17337 8.012 1.25e-13 ***
#> y1_mean 0.06308 0.06038 1.045 0.29749
#> y2_diff 0.13111 0.04081 3.213 0.00155 **
#> y2_mean -0.04300 0.05654 -0.761 0.44789
#> ---
#> Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
#>
#> Residual standard error: 1.108 on 184 degrees of freedom
#> Multiple R-squared: 0.06089, Adjusted R-squared: 0.04557
#> F-statistic: 3.976 on 3 and 184 DF, p-value: 0.00893The output of the model includes estimates of the regression coefficients, standard errors, and p-values for the association between the two outcome variables.
Alternative Specifications
If desired, one can manually prepare the data using
discord_data() and then fit separate models for the
individual-level, between-pair, and within-pair effects. This approach
provides more flexibility in specifying the models and allows for a
deeper understanding of the different components of the association.
data_df <- discord_data(
data = data_demo,
outcome = "y1",
predictors = "y2",
id = "id",
sex = NULL,
race = NULL,
demographics = "none",
pair_identifiers = c("_1", "_2")
)
summary(data_df)
#> id y1_1 y1_2 y1_diff
#> Min. : 1.00 Min. :-1.457 Min. :-3.3672 Min. :0.003215
#> 1st Qu.: 47.75 1st Qu.: 1.643 1st Qu.: 0.1777 1st Qu.:0.707853
#> Median : 94.50 Median : 2.723 Median : 1.1478 Median :1.311807
#> Mean : 94.50 Mean : 2.679 Mean : 1.1538 Mean :1.524696
#> 3rd Qu.:141.25 3rd Qu.: 3.882 3rd Qu.: 2.1351 3rd Qu.:2.207785
#> Max. :188.00 Max. : 6.154 Max. : 4.9615 Max. :5.157103
#> y1_mean y2_1 y2_2 y2_diff
#> Min. :-1.700 Min. :-2.109 Min. :-2.959 Min. :-3.9241
#> 1st Qu.: 1.006 1st Qu.: 1.291 1st Qu.: 0.574 1st Qu.:-0.4878
#> Median : 1.912 Median : 2.364 Median : 1.505 Median : 0.8402
#> Mean : 1.916 Mean : 2.478 Mean : 1.682 Mean : 0.7951
#> 3rd Qu.: 2.884 3rd Qu.: 3.602 3rd Qu.: 2.775 3rd Qu.: 2.0682
#> Max. : 5.053 Max. : 6.788 Max. : 6.783 Max. : 6.9832
#> y2_mean
#> Min. :-1.154
#> 1st Qu.: 1.077
#> Median : 1.936
#> Mean : 2.080
#> 3rd Qu.: 2.911
#> Max. : 6.119
lm_ind <- lm(y1_1 ~ y2_1, data = data_df)
summary(lm_ind)
#>
#> Call:
#> lm(formula = y1_1 ~ y2_1, data = data_df)
#>
#> Residuals:
#> Min 1Q Median 3Q Max
#> -4.1295 -0.9876 0.0817 1.1612 3.7977
#>
#> Coefficients:
#> Estimate Std. Error t value Pr(>|t|)
#> (Intercept) 2.30725 0.18995 12.146 <2e-16 ***
#> y2_1 0.14987 0.06282 2.386 0.018 *
#> ---
#> Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
#>
#> Residual standard error: 1.493 on 186 degrees of freedom
#> Multiple R-squared: 0.0297, Adjusted R-squared: 0.02448
#> F-statistic: 5.692 on 1 and 186 DF, p-value: 0.01804
lm_ind2 <- lm(y1_2 ~ y2_2, data = data_df)
summary(lm_ind2)
#>
#> Call:
#> lm(formula = y1_2 ~ y2_2, data = data_df)
#>
#> Residuals:
#> Min 1Q Median 3Q Max
#> -4.4598 -0.9950 -0.0377 0.9567 3.3692
#>
#> Coefficients:
#> Estimate Std. Error t value Pr(>|t|)
#> (Intercept) 0.87682 0.14193 6.178 4e-09 ***
#> y2_2 0.16466 0.05788 2.845 0.00494 **
#> ---
#> Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
#>
#> Residual standard error: 1.416 on 186 degrees of freedom
#> Multiple R-squared: 0.0417, Adjusted R-squared: 0.03655
#> F-statistic: 8.094 on 1 and 186 DF, p-value: 0.004939
lm_between <- lm(y1_mean ~ y2_mean, data = data_df)
summary(lm_between)
#>
#> Call:
#> lm(formula = y1_mean ~ y2_mean, data = data_df)
#>
#> Residuals:
#> Min 1Q Median 3Q Max
#> -3.7107 -0.8579 0.0237 0.9435 3.3296
#>
#> Coefficients:
#> Estimate Std. Error t value Pr(>|t|)
#> (Intercept) 1.5642 0.1713 9.131 <2e-16 ***
#> y2_mean 0.1692 0.0675 2.507 0.013 *
#> ---
#> Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
#>
#> Residual standard error: 1.346 on 186 degrees of freedom
#> Multiple R-squared: 0.03268, Adjusted R-squared: 0.02748
#> F-statistic: 6.284 on 1 and 186 DF, p-value: 0.01304
lm_within <- lm(y1_diff ~ y1_mean + y2_diff + y2_mean, data = data_df)
summary(lm_within)
#>
#> Call:
#> lm(formula = y1_diff ~ y1_mean + y2_diff + y2_mean, data = data_df)
#>
#> Residuals:
#> Min 1Q Median 3Q Max
#> -1.8511 -0.8476 -0.1705 0.6317 3.6083
#>
#> Coefficients:
#> Estimate Std. Error t value Pr(>|t|)
#> (Intercept) 1.38903 0.17337 8.012 1.25e-13 ***
#> y1_mean 0.06308 0.06038 1.045 0.29749
#> y2_diff 0.13111 0.04081 3.213 0.00155 **
#> y2_mean -0.04300 0.05654 -0.761 0.44789
#> ---
#> Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
#>
#> Residual standard error: 1.108 on 184 degrees of freedom
#> Multiple R-squared: 0.06089, Adjusted R-squared: 0.04557
#> F-statistic: 3.976 on 3 and 184 DF, p-value: 0.00893Conclusion
This vignette demonstrates how {BGmisc} and discord enable researchers to perform discordant-kinship analyses starting from simple family data. There’s no need for pre-constructed kinship links or specialized datasets like the NLSY—just basic family identifiers are sufficient to generate kinship structures and apply powerful behavior genetic models.