Full Data Workflow
S. Mason Garrison
Source:vignettes/full_data_workflow.Rmd
full_data_workflow.RmdIntroduction
This vignette provides a complete workflow for conducting discordant-kinship regression using the {discord} package. We encourage you to read the accompanying paper (Garrison et al., 2025) for a full explanation of the methods and their applications. We show how to transform data into a format suitable for analysis, then walk through standard OLS regression (for comparison) and discordant-kinship regression.
The tutorial covers three common data structures (wide, long, and pedigree formats) and uses simulated data for reproducibility. You can adapt our R-based analysis pipeline to your own data by substituting your variables and kinship links.
Data Structures and Preparation
Your data likely exists in one of three common formats: wide, long,
or pedigree. Regardless of format, the goal is to create wide-format
data where each row represents a kinship pair. Variables for each member
of the pair are distinguished by suffixes (e.g., _1 and
_2). Understanding your data structure determines which
transformation steps you’ll need. Each subsection below outlines how to
handle these formats.
First, load the necessary packages:
# For easy data manipulation
library(dplyr)
library(tidyr)
# For kinship linkages
library(NlsyLinks)
# For discordant-kinship regression
library(discord)
# To clean data frames
library(janitor)
library(broom)
# For pipe
library(magrittr)
# For pedigree data manipulation
library(BGmisc)
# For pedigree plotting
library(ggpedigree)
library(ggplot2)Once you load the necessary packages, feel free to skip to the relevant subsection for your data type.
Wide Format Data
Wide format is the most direct structure for our analysis. Each row
represents one kinship pair, with variables appearing twice—once for
each member of the pair—using suffixes to distinguish the two
individuals. For example, columns like age_s1 and
age_s2, where _s1 and _s2
identify which sibling’s data appears in each column.
The simulated sibling data below contains five sibling pairs with variables for ID, age, height, and weight for each sibling.
df_wide <- data.frame(
pid = 1:5,
id_s1 = c(101, 201, 301, 401, 501),
id_s2 = c(102, 202, 302, 402, 502),
age_s1 = c(30, 27, 40, 36, 30),
age_s2 = c(28, 25, 38, 35, 28),
height_s1 = c(175, 160, 180, 170, 165),
height_s2 = c(170, 162, 178, 172, 168),
weight_s1 = c(70, 60, 80, 75, 65),
weight_s2 = c(68, 62, 78, 74, 66)
)
df_wide %>%
slice(1:5) %>%
knitr::kable()| pid | id_s1 | id_s2 | age_s1 | age_s2 | height_s1 | height_s2 | weight_s1 | weight_s2 |
|---|---|---|---|---|---|---|---|---|
| 1 | 101 | 102 | 30 | 28 | 175 | 170 | 70 | 68 |
| 2 | 201 | 202 | 27 | 25 | 160 | 162 | 60 | 62 |
| 3 | 301 | 302 | 40 | 38 | 180 | 178 | 80 | 78 |
| 4 | 401 | 402 | 36 | 35 | 170 | 172 | 75 | 74 |
| 5 | 501 | 502 | 30 | 28 | 165 | 168 | 65 | 66 |
In this example, pid is a unique identifier for each
sibling pair. Specifically, each row is anchored by a pair identifier.
Variables ending in _s1 refer to one sibling, while those
ending in _s2 refer to the other. In our case, the starting
dataset is already arranged so that the older sibling is
_s1 and the younger is _s2. Nevertheless, this
ordering will be adjusted later based on the outcome variable. If your
data already has this structure, you can skip ahead to the “Ordering and
Derived Variables” section.
Long Format Data
Long format structures data with one row per individual. In other words, each person appears in their own row, with a pair-identifier column linking siblings. This format is common in many datasets (such as the NLSY), but it requires transformation for our analysis.
We can demonstrate long format by reshaping our wide data from above
into long format using pivot_longer() from the {tidyr}
package:
df_long <- df_wide %>%
tidyr::pivot_longer(
cols = -pid, # keep the dyad identifier intact
names_to = c(".value", "sibling"), # split base names and the sibling marker
names_sep = "_s" # original suffix delimiter in column names
)
df_long %>%
slice(1:10) %>%
knitr::kable()| pid | sibling | id | age | height | weight |
|---|---|---|---|---|---|
| 1 | 1 | 101 | 30 | 175 | 70 |
| 1 | 2 | 102 | 28 | 170 | 68 |
| 2 | 1 | 201 | 27 | 160 | 60 |
| 2 | 2 | 202 | 25 | 162 | 62 |
| 3 | 1 | 301 | 40 | 180 | 80 |
| 3 | 2 | 302 | 38 | 178 | 78 |
| 4 | 1 | 401 | 36 | 170 | 75 |
| 4 | 2 | 402 | 35 | 172 | 74 |
| 5 | 1 | 501 | 30 | 165 | 65 |
| 5 | 2 | 502 | 28 | 168 | 66 |
Notice how each individual now occupies their own row. The
sibling column indicating whether each sibling was
originally labeled 1 or 2. The
pid column still identifies which individuals form a
pair.
Suppose your data does not already have a unique identifier for each kinship pair. In that case, you may need to construct one based on the available information or download one from a kinship linkage resource. Ideally, you will also have information about the type of kinship (e.g., full siblings, half-siblings, cousins) and whether they were raised together or apart. (More on how to do this below for pedigree data.)
Converting Long to Wide
To convert long format data for discordant-kinship analysis, use
pivot_wider() from the {tidyr} package:
df_long2wide <- df_long %>%
tidyr::pivot_wider(
names_from = sibling, # the column that indicates the sibling number
values_from = c(id, age, height, weight), # variables to spread into paired columns
names_sep = "_s" # ensures id_s1, id_s2,etc
)
df_long2wide %>%
slice(1:5) %>%
knitr::kable()| pid | id_s1 | id_s2 | age_s1 | age_s2 | height_s1 | height_s2 | weight_s1 | weight_s2 |
|---|---|---|---|---|---|---|---|---|
| 1 | 101 | 102 | 30 | 28 | 175 | 170 | 70 | 68 |
| 2 | 201 | 202 | 27 | 25 | 160 | 162 | 60 | 62 |
| 3 | 301 | 302 | 40 | 38 | 180 | 178 | 80 | 78 |
| 4 | 401 | 402 | 36 | 35 | 170 | 172 | 75 | 74 |
| 5 | 501 | 502 | 30 | 28 | 165 | 168 | 65 | 66 |
The reshaped data now reproduces our original wide format. Each
analytic variable appears twice with _s1 and
_s2 suffixes, and the pair identifier pid
anchors each row.
Using existing kinship links
As mentioned before, if your long-format data lacks pair identifiers, you’ll need to create them using family IDs, household identifiers, or external kinship resources. This process can be straightforward if you have access to family or household identifiers, and are confident in the kinship relationships. However, it can be more complex if the relationships are not clearly defined or if there are multiple types of kinship (e.g., siblings, cousins, half-siblings) in your data.
For NLSY data specifically, the {NlsyLinks} package provides validated kinship links for the vast majority of dyads in the NLSY79 and NLSY97 cohorts. These links are provided in wide format, and can be used to left-join with the long-format data that the NLSY info website provides. See the {NlsyLinks} documentation for more details on how to access and use these datasets, or the reproducible source code for the analyses in the primary paper (Garrison et al., 2025).
library(NlsyLinks)
data(Links79PairExpanded)
Links79PairExpanded %>%
arrange(ExtendedID) %>%
filter(RelationshipPath == "Gen1Housemates" & RFull == 0.5) %>%
select(
ExtendedID,
SubjectTag_S1, SubjectTag_S2,
RelationshipPath, RFull, IsMz,
EverSharedHouse
) %>% # used to make the table
slice_head(n = 5) %>%
knitr::kable()| ExtendedID | SubjectTag_S1 | SubjectTag_S2 | RelationshipPath | RFull | IsMz | EverSharedHouse |
|---|---|---|---|---|---|---|
| 3 | 300 | 400 | Gen1Housemates | 0.5 | No | TRUE |
| 5 | 500 | 600 | Gen1Housemates | 0.5 | No | TRUE |
| 13 | 1300 | 1400 | Gen1Housemates | 0.5 | No | TRUE |
| 17 | 1700 | 1800 | Gen1Housemates | 0.5 | No | TRUE |
| 20 | 2000 | 2100 | Gen1Housemates | 0.5 | No | TRUE |
As you can see, this dataset includes an extended family identifier
(ExtendedID), individual IDs for each sibling in the pair
(R0000100 and R0000200), and their
RelationshipPath (RelationshipPath = “Gen1Housemates” and
genetic relatedness (RFull = 0.5). You can merge this
dataset with your long-format data using the individual IDs.
At this stage the long‑origin data are indistinguishable from the
wide-origin wide example. Each analytic variable now appears exactly
twice—once for _s1 and once for _s2—and the
dyad identifier pid continues to anchor the row. You can
proceed directly to ordering and construction of *_mean and
*_diff with discord_data().
Pedigree Data
Pedigree data is common in genetic and family studies, where detailed family trees are available. It is long format data that provides rich information about familial relationships, such as (mother ID, father ID, spouse ID). Our research team has developed specialized tools in the {BGmisc} package to extract kinship links and transform these pedigree structures into a wide format suitable for analysis. Below is an example of how such a dataset might look in both tabular and graphical forms:
data(potter)
ggpedigree(potter, config = list(
label_method = "geom_text",
label_nudge_y = .25,
focal_fill_personID = 7,
focal_fill_include = TRUE,
focal_fill_force_zero = TRUE,
focal_fill_na_value = "grey50",
focal_fill_low_color = "darkred",
focal_fill_high_color = "gold",
focal_fill_mid_color = "orange",
focal_fill_scale_midpoint = .65,
focal_fill_component = "additive",
focal_fill_method = "steps", #
# focal_fill_method = "viridis_c",
focal_fill_use_log = FALSE,
focal_fill_n_breaks = 10,
sex_color_include = F,
focal_fill_legend_title = "Genetic Relatives \nof Harry Potter"
)) +
labs(title = "Potter Pedigree Plot") +
theme(legend.position = "right")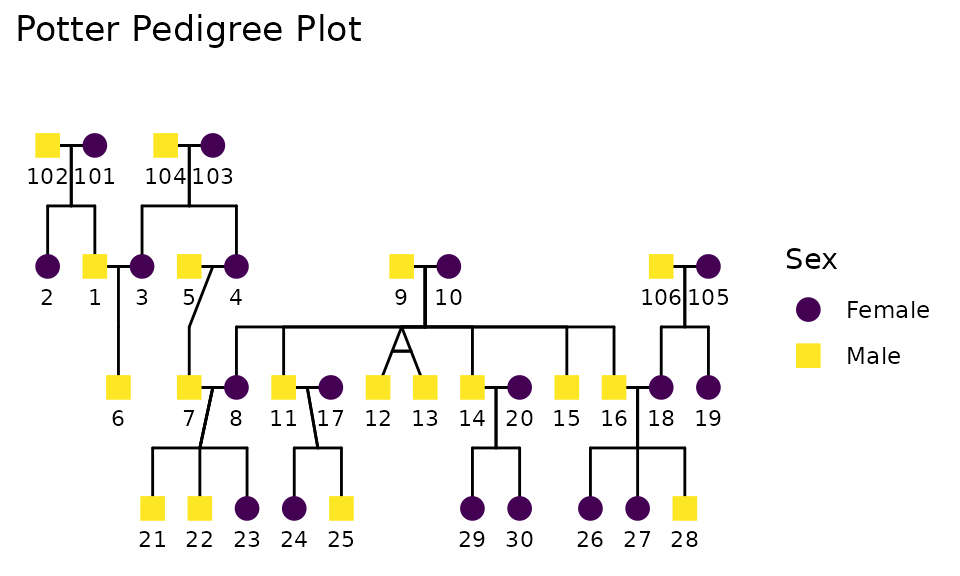
The pedigree tree above illustrates the family relationships among individuals in the Potter dataset. Each node represents an individual, and the lines connecting them indicate familial relationships such as parent-child and sibling connections.
To work with pedigree data, we first need to convert it into a long
format data frame. The following code extracts relevant columns from the
potter dataset and adds a synthetic variable
(x_var) for demonstration purposes:
data(potter)
df_ped <- potter %>%
as.data.frame() %>%
select(
personID, sex, famID, momID, dadID, spouseID,
twinID, zygosity
) %>%
mutate(x_var = round(rnorm(nrow(.), mean = 0, sd = 1), digits = 2))
df_ped %>%
slice(1:5) %>%
knitr::kable(digits = 2)| personID | sex | famID | momID | dadID | spouseID | twinID | zygosity | x_var |
|---|---|---|---|---|---|---|---|---|
| 1 | 1 | 1 | 101 | 102 | 3 | NA | NA | -1.40 |
| 2 | 0 | 1 | 101 | 102 | NA | NA | NA | 0.26 |
| 3 | 0 | 1 | 103 | 104 | 1 | NA | NA | -2.44 |
| 4 | 0 | 1 | 103 | 104 | 5 | NA | NA | -0.01 |
| 5 | 1 | 1 | NA | NA | 4 | NA | NA | 0.62 |
As you can see, each individual is represented in a separate row, with columns for their unique identifier, mother’s identifier, father’s identifier, and other relevant variables. The {BGmisc} package provides functions to compute kinship matrices from pedigree data. The package is available on CRAN and GitHub. The package ( and its documentation ) can be found at: https://CRAN.R-project.org/package=BGmisc as well as it’s accompanying Journal of Open Source Software paper: https://joss.theoj.org/papers/10.21105/joss.06203
Computing Kinship Matrices
To transform this data into a wide format suitable for discordant-kinship regression, we need to create kinship links based on the pedigree information. To extract the necessary kinship information, we need to compute two matrices: the additive genetic relatedness matrix (add) and the shared environment matrix (cn). Other matrices, such as mitochondrial, can also be computed if needed.
The ped2add() function computes the additive genetic
relatedness matrix, which quantifies the genetic similarity between
individuals based on their pedigree information. The
ped2cn() function computes the shared environment matrix,
which indicates whether individuals were raised in the same environment
(1) or different environments (0).
The resulting matrices are symmetric, with diagonal elements representing self-relatedness (1.0). The off-diagonal elements represent the relatedness between pairs of individuals, with values ranging from 0 (no relatedness) to 0.5 (full siblings) to 1 (themselves).
Creating Wide-Format Kinship Pairs
We convert the component matrices into a wide-form dataframe of kin
pairs using com2links(). Self-pairs and duplicate entries
are removed.
df_links <- com2links(
writetodisk = FALSE,
ad_ped_matrix = add,
cn_ped_matrix = cn,
drop_upper_triangular = TRUE
) %>%
filter(ID1 != ID2)
df_links %>%
slice(1:5) %>%
knitr::kable(digits = 3)| ID1 | ID2 | addRel | cnuRel |
|---|---|---|---|
| 1 | 2 | 0.50 | 1 |
| 3 | 4 | 0.50 | 1 |
| 1 | 6 | 0.50 | 0 |
| 2 | 6 | 0.25 | 0 |
| 3 | 6 | 0.50 | 0 |
As you can see, the df_links data frame contains pairs
of individuals (ID1 and ID2) along with their additive genetic
relatedness (addRel) and shared environment status
(cnuRel). These data are in wide format, with each row
representing a unique pair of individuals.
Further, we can tally the number of pairs by relatedness and shared environment to understand the composition of the dataset.
| addRel | cnuRel | n |
|---|---|---|
| 0.0625 | 0 | 3 |
| 0.1250 | 0 | 47 |
| 0.2500 | 0 | 104 |
| 0.5000 | 0 | 50 |
| 0.5000 | 1 | 32 |
This table shows the number of kinship pairs for each combination of genetic relatedness and shared environment status. Although the discord regression models can be used with any kin group, it is most interpretable when there is a single kinship group.
Feel free to merge this df_links data frame with your
pedigree data to include additional variables for each individual in the
pair.
The following optional subsection demonstrates how to simulate outcome and predictor variables for these kinship pairs.
Simulating Outcome and Predictor Variables
To simulate outcome and predictor variables for our kinship pairs, we
can use the kinsim() function from the {discord} package.
This function allows us to generate synthetic data based on specified
genetic and environmental parameters.
For demonstration, we’ll focus on first cousins raised in separate homes from our pedigree data.
df_cousin <- df_links %>%
filter(addRel == .125) %>% # only cousins %>%
filter(cnuRel == 0) # only kin raised in separate homeskinsim allows us to generate synthetic data based on
specified genetic and environmental parameters or relatedness vectors.
Here, we simulate data for first cousins raised apart (addRel = 0.125,
cnuRel = 0) using the df_links data frame we created
earlier.
set.seed(2024)
df_synthetic <- discord::kinsim(
mu_all = c(1, 1), # means
cov_a = .5,
cov_c = .1, #
cov_e = .3,
c_vector = rep(df_cousin$cnuRel, 3),
r_vector = rep(df_cousin$addRel, 3)
)I now have a synthetic dataset containing pairs of first cousins
raised apart, with simulated values for weight and height. Each row
represents a unique pair of cousins, with variables for each cousin
distinguished by the _1 and _2 suffixes. This
data is designed such that our weight and height variables have genetic
(cov_a=.5) and environmental correlations (cov_c=.1, cov_e=.3). By
default, univariate ACE values are 1/3 genetic, 1/3 shared environment,
and 1/3 unique environment. Also by default, kinsim()
generates variables named y1 and y2 for the
first and second variables, respectively. We can rename these to more
meaningful names like weight and height for
clarity.
df_synthetic <- df_synthetic %>%
select(
pid = id,
r,
weight_s1 = y1_1,
weight_s2 = y1_2,
height_s1 = y2_1,
height_s2 = y2_2
) %>%
mutate( # simulates age such that the 2nd sibling is between 1 and 5 years younger.
age_s1 = round(rnorm(nrow(.), mean = 30, sd = 5), digits = 0),
age_s2 = age_s1 - sample(1:5, nrow(.), replace = TRUE)
)
df_synthetic %>%
slice(1:5) %>%
knitr::kable(digits = 2)| pid | r | weight_s1 | weight_s2 | height_s1 | height_s2 | age_s1 | age_s2 |
|---|---|---|---|---|---|---|---|
| 1 | 0.12 | 2.09 | -1.30 | 4.54 | -0.66 | 31 | 30 |
| 2 | 0.12 | 1.21 | 0.40 | 1.20 | 0.26 | 32 | 28 |
| 3 | 0.12 | 1.03 | 2.53 | 2.76 | 0.58 | 29 | 27 |
| 4 | 0.12 | 1.46 | 4.92 | 0.93 | -0.67 | 30 | 29 |
| 5 | 0.12 | 2.69 | 2.78 | 0.95 | -0.54 | 23 | 18 |
This synthetic dataset now contains pairs of first cousins raised
apart, with simulated values for weight and height, as well as age. Each
row represents a unique pair of cousins, with variables for each cousin
distinguished by the _s1 and _s2 suffixes.
Ordering and Derived Variables
Now that we have our data in wide format, we can proceed to order the pairs and create the derived variables needed for discordant-kinship regression. The key steps are:
-
Ordering the pairs: We need to ensure that within
each pair, the individual with the higher outcome value is consistently
labeled as
_1and the other as_2. This ordering is to ensure that we can extract meaningful difference scores. -
Creating derived variables: We will create
*_meanand*_diff*variables for both the outcome and predictor variables. The*_meanvariable represents the average of the two individuals’ values, while the*_diffvariable represents the difference between the two individuals’ values (i.e.,_1 - _2).
These steps can be accomplished using the discord_data()
function from the {discord} package. This function takes care of
ordering the pairs and creating the derived variables, ensuring that the
data is ready for analysis. When using discord_data(), you
will need to specify the outcome variable, predictor variables, and the
identifiers for the two members of each pair.
Below we default to pedigree data of cousins for reproducibility. Replace with your chosen source as needed.
# CHOOSE ONE based on your path
# source_wide <- df_wide
# source_wide <- df_long2wide
source_wide <- df_synthetic # if you followed the pedigree pathNow call discord_data() specifying the outcome and
predictor present for both siblings. Here we use weight as
the outcome and height and age as the
predictors, with the _s1 / _s2 suffix
convention.
df_discord_weight <- discord::discord_data(
data = source_wide,
outcome = "weight",
predictors = c("height", "age"),
demographics = "none",
pair_identifiers = c("_s1", "_s2"),
id = "pid" # or "famID"
)
df_discord_weight %>%
slice(1:5) %>%
knitr::kable(digits = 2, caption = "Transformed data ready for discordant-kinship regression")| id | weight_1 | weight_2 | weight_diff | weight_mean | height_1 | height_2 | height_diff | height_mean | age_1 | age_2 | age_diff | age_mean |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2.09 | -1.30 | 3.39 | 0.39 | 4.54 | -0.66 | 5.20 | 1.94 | 31 | 30 | 1 | 30.5 |
| 2 | 1.21 | 0.40 | 0.81 | 0.80 | 1.20 | 0.26 | 0.94 | 0.73 | 32 | 28 | 4 | 30.0 |
| 3 | 2.53 | 1.03 | 1.51 | 1.78 | 0.58 | 2.76 | -2.18 | 1.67 | 27 | 29 | -2 | 28.0 |
| 4 | 4.92 | 1.46 | 3.46 | 3.19 | -0.67 | 0.93 | -1.60 | 0.13 | 29 | 30 | -1 | 29.5 |
| 5 | 2.78 | 2.69 | 0.09 | 2.74 | -0.54 | 0.95 | -1.49 | 0.21 | 18 | 23 | -5 | 20.5 |
Understanding the Transformation
Let’s examine what discord_data() did to our
variables:
# Show original data for first 3 pairs
source_wide %>%
slice(1:3) %>%
select(pid, weight_s1, weight_s2, height_s1, height_s2) %>%
knitr::kable(
digits = 2,
caption = "Original data: siblings not yet ordered by outcome"
)| pid | weight_s1 | weight_s2 | height_s1 | height_s2 |
|---|---|---|---|---|
| 1 | 2.09 | -1.30 | 4.54 | -0.66 |
| 2 | 1.21 | 0.40 | 1.20 | 0.26 |
| 3 | 1.03 | 2.53 | 2.76 | 0.58 |
df_discord_weight %>%
select(
id,
weight_1, weight_2, weight_mean, weight_diff,
height_1, height_2, height_mean, height_diff
) %>%
slice(1:3) %>%
knitr::kable(
digits = 2,
caption = "After discord_data(): siblings ordered so weight_1 >= weight_2"
)| id | weight_1 | weight_2 | weight_mean | weight_diff | height_1 | height_2 | height_mean | height_diff |
|---|---|---|---|---|---|---|---|---|
| 1 | 2.09 | -1.30 | 0.39 | 3.39 | 4.54 | -0.66 | 1.94 | 5.20 |
| 2 | 1.21 | 0.40 | 0.80 | 0.81 | 1.20 | 0.26 | 0.73 | 0.94 |
| 3 | 2.53 | 1.03 | 1.78 | 1.51 | 0.58 | 2.76 | 1.67 | -2.18 |
Notice several important patterns in this output. First, the ordering changes depending on which sibling has the higher outcome value:
- If
weight_s1 > weight_s2in the original data, sibling 1 becomes_1in the output - If
weight_s2 > weight_s1in the original data, the siblings are swapped: sibling 2 becomes_1 - The predictor values (height) are reordered accordingly to stay matched with the correct sibling
The sibling with higher weight becomes _1 and the
sibling with lower weight becomes _2. This ordering ensures
that weight_diff (calculated as
weight_1 - weight_2) is always non-negative. In the case of
ties, discord data randomly assigned one sibling as _1 and
perserves that ordering throughout the dataset. Extremely motivated
readers can dive into the discord source code for exactly how this
calculation is implemented
Second, the mean scores represent each pair’s average weight. For
example, weight_mean equals the average of
weight_1 and weight_2. These means capture
between-family variation, reflecting differences across sibling pairs so
that we can compare families to one another.
Third, the predictor differences can be positive or negative. Even though weight differences are always non-negative (by construction), height differences vary. The taller sibling might be the heavier one, or the shorter sibling might be heavier. This variation is what we’ll test in our regression.
This structure lets us ask a key question: within sibling pairs, does the sibling with more of the predictor also have more of the outcome? If the taller sibling is systematically heavier even after controlling for family background, that suggests height may causally influence weight. If not, the association likely reflects familial confounding.
Data Selection for Standard OLS Regression
For comparison, we also need to prepare data for a standard OLS regression. When running a standard OLS regression, researchers typically select one sibling per family to avoid non-independence. Common approaches include:
- First-born sibling: The child born first in the family
- Oldest sibling: The sibling with the highest age at time of measurement
- Random selection: Randomly choosing one sibling from each pair
This selection is typically fixed based on a demographic characteristic (birth order, age) and remains constant regardless of which outcome variable you’re analyzing. In contrast, discordant-kinship regression dynamically orders siblings based on the outcome variable. Thus each analysis may involve different sibling orderings.
Analyzing the Data
With our data prepared, we can now perform both standard OLS regression and discordant-kinship regression to compare results.
Standard OLS Regression
First, let’s run a standard OLS regression using the original wide-format data. This will serve as a baseline for comparison with the family-based analyses.
For our baseline analysis, we select one sibling from each pair. In
the original dataset, s1 was defined as the older sibling, so we can
simply choose the sibling labeled _s1. But, another way to
select the oldest sibling is to use the discord_data()
function with the outcome argument set to the age variable.
This approach will ensure that _1 is always the oldest
sibling within the pair. In the case of ties, discord data randomly
assigned one sibling as _1. This assignment is preserved
thru the entire data management process.
df_for_ols <- df_synthetic %>%
dplyr::select(
id = pid,
weight = weight_s1,
height = height_s1,
age = age_s1
)
df_discord_age <- discord::discord_data(
data = source_wide,
outcome = "age",
predictors = c("height", "weight"),
demographics = "none",
pair_identifiers = c("_s1", "_s2"),
id = "pid" # or "famID" if you followed the pedigree path
)The dataframes df_for_ols and
df_discord_age now both contain one sibling per pair,
selected based on age. We can verify that they contain the same
individuals:
df_discord_age %>%
select(
id, age_1, age_2,
age_mean, age_diff, weight_1, height_1
) %>%
slice(1:5) %>%
knitr::kable(caption = "One sibling per pair for OLS regression, selecting by age", digits = 2)| id | age_1 | age_2 | age_mean | age_diff | weight_1 | height_1 |
|---|---|---|---|---|---|---|
| 1 | 31 | 30 | 30.5 | 1 | 2.09 | 4.54 |
| 2 | 32 | 28 | 30.0 | 4 | 1.21 | 1.20 |
| 3 | 29 | 27 | 28.0 | 2 | 1.03 | 2.76 |
| 4 | 30 | 29 | 29.5 | 1 | 1.46 | 0.93 |
| 5 | 23 | 18 | 20.5 | 5 | 2.69 | 0.95 |
df_for_ols %>%
select(id, age, weight, height) %>%
slice(1:5) %>%
knitr::kable(caption = "One sibling per pair for OLS regression using original wide data", digits = 2)| id | age | weight | height |
|---|---|---|---|
| 1 | 31 | 2.09 | 4.54 |
| 2 | 32 | 1.21 | 1.20 |
| 3 | 29 | 1.03 | 2.76 |
| 4 | 30 | 1.46 | 0.93 |
| 5 | 23 | 2.69 | 0.95 |
As you can see, df_for_ols and
df_discord_age both resulted in the same individuals being
selected for analysis. The df_discord_age dataset was
created by ordering siblings based on age, ensuring that _1
is always the older sibling. The df_for_ols dataset
directly selects the older sibling from the original wide data.
By selecting one sibling per pair, we’re analyzing the same number of observations as we have kinship pairs. This conrespondses with the standard individual-level approach used in most research:
ols_model <- lm(weight ~ height + age, data = df_for_ols)
stargazer::stargazer(ols_model,
type = "html", ci = TRUE,
digits = 3, single.row = TRUE, title = "Standard OLS Regression Results"
)| Dependent variable: | |
| weight | |
| height | 0.287*** (0.134, 0.440) |
| age | -0.073** (-0.129, -0.017) |
| Constant | 2.788*** (1.081, 4.495) |
| Observations | 141 |
| R2 | 0.125 |
| Adjusted R2 | 0.112 |
| Residual Std. Error | 1.573 (df = 138) |
| F Statistic | 9.857*** (df = 2; 138) |
| Note: | p<0.1; p<0.05; p<0.01 |
stargazer::stargazer(ols_model,
type = "latex", ci = TRUE,
digits = 3, single.row = TRUE, title = "Standard OLS Regression Results"
)This standard regression shows associations between our variables while controlling only for measured covariates.
Discordant Between-Family Regression
Next, we run a between-family regression using the discordant data. This model regresses the outcome mean on predictor means, capturing between-family variation:
between_model <- lm(
weight_mean ~ height_mean + age_mean,
data = df_discord_weight
)
stargazer::stargazer(between_model,
type = "html", ci = TRUE,
digits = 3, single.row = TRUE, title = "Between-Family Regression Results"
)| Dependent variable: | |
| weight_mean | |
| height_mean | 0.199** (0.049, 0.350) |
| age_mean | -0.044* (-0.089, 0.001) |
| Constant | 2.064*** (0.759, 3.368) |
| Observations | 141 |
| R2 | 0.067 |
| Adjusted R2 | 0.054 |
| Residual Std. Error | 1.284 (df = 138) |
| F Statistic | 4.970*** (df = 2; 138) |
| Note: | p<0.1; p<0.05; p<0.01 |
This between-family regression captures how differences in average height and age across families relate to average weight. However, it does not account for within-family differences, which is where discordant-kinship regression comes in.
It is important to note that the between-family regression results are not directly comparable to the standard OLS regression results. The between-family model uses mean scores, which represent family-level averages, while the OLS model uses individual-level data. Therefore, the coefficients from these two models may differ in magnitude and interpretation. But, they both provide useful information about the relationships among the variables.
Discordant-Kinship Regression
Now we return to our prepared data to run discordant-kinship
regression. We can specify the models manually or use the
discord_regression() convenience function. Both approaches
produce identical results.
The discordant model regresses the outcome difference on outcome mean, predictor means, and predictor differences:
When fitting the discordant regression model, the coefficient for each
*_mean variable captures the between-family effect, while
the coefficient for each *_diff variable captures the
within-family effect. The within-family effects are of particular
interest, as they provide insight into how differences between siblings
relate to differences in the outcome, controlling for shared family
background. Below you can see how this model is specified manually:
discord_model_manual <- lm(
weight_diff ~ weight_mean + height_mean + height_diff + age_mean + age_diff,
data = df_discord_weight
)
tidy(discord_model_manual, conf.int = TRUE) %>%
knitr::kable(digits = 3, caption = "Discordant Regression (Manual)")| term | estimate | std.error | statistic | p.value | conf.low | conf.high |
|---|---|---|---|---|---|---|
| (Intercept) | 0.500 | 0.528 | 0.948 | 0.345 | -0.544 | 1.544 |
| weight_mean | -0.043 | 0.065 | -0.660 | 0.511 | -0.171 | 0.086 |
| height_mean | 0.041 | 0.060 | 0.680 | 0.498 | -0.078 | 0.159 |
| height_diff | 0.141 | 0.043 | 3.254 | 0.001 | 0.055 | 0.226 |
| age_mean | 0.031 | 0.018 | 1.742 | 0.084 | -0.004 | 0.066 |
| age_diff | -0.026 | 0.024 | -1.092 | 0.277 | -0.074 | 0.021 |
stargazer::stargazer(between_model, discord_model_manual,
type = "html", ci = TRUE,
digits = 3, single.row = TRUE, title = "Between-Family and Discordant Regression Results"
)| Dependent variable: | ||
| weight_mean | weight_diff | |
| (1) | (2) | |
| weight_mean | -0.043 (-0.170, 0.084) | |
| height_mean | 0.199** (0.049, 0.350) | 0.041 (-0.077, 0.158) |
| height_diff | 0.141*** (0.056, 0.225) | |
| age_mean | -0.044* (-0.089, 0.001) | 0.031* (-0.004, 0.066) |
| age_diff | -0.026 (-0.074, 0.021) | |
| Constant | 2.064*** (0.759, 3.368) | 0.500 (-0.534, 1.534) |
| Observations | 141 | 141 |
| R2 | 0.067 | 0.102 |
| Adjusted R2 | 0.054 | 0.069 |
| Residual Std. Error | 1.284 (df = 138) | 0.978 (df = 135) |
| F Statistic | 4.970*** (df = 2; 138) | 3.060** (df = 5; 135) |
| Note: | p<0.1; p<0.05; p<0.01 | |
Note that we are using the df_discord_weight dataset created earlier
with discord_data(). This dataset is ordered such that
within a pair, the sibling with the higher weight is always
_1. This ordering ensures that weight_diff is
always non-negative, which is a key feature of discordant-kinship
regression.
Using the Helper Function
Alternatively, we can use the discord_regression()
function, which simplifies the process:
By default, discord_regression() orders siblings by the
outcome variable, creates the necessary derived variables, and fits the
discordant regression model in one step. This function also allows for
easy inclusion of demographic covariates if needed. See the function
documentation for more details on these options, as well as a vignette
focused on demographic controls.
discord_model <- discord_regression(
data = source_wide,
outcome = "weight",
predictors = c("height", "age"),
demographics = "none",
sex = NULL,
race = NULL,
pair_identifiers = c("_s1", "_s2"),
id = "pid"
)
tidy(discord_model, conf.int = TRUE) %>%
knitr::kable(digits = 3, caption = "Discordant Regression Results")| term | estimate | std.error | statistic | p.value | conf.low | conf.high |
|---|---|---|---|---|---|---|
| (Intercept) | 0.500 | 0.528 | 0.948 | 0.345 | -0.544 | 1.544 |
| weight_mean | -0.043 | 0.065 | -0.660 | 0.511 | -0.171 | 0.086 |
| height_diff | 0.141 | 0.043 | 3.254 | 0.001 | 0.055 | 0.226 |
| age_diff | -0.026 | 0.024 | -1.092 | 0.277 | -0.074 | 0.021 |
| height_mean | 0.041 | 0.060 | 0.680 | 0.498 | -0.078 | 0.159 |
| age_mean | 0.031 | 0.018 | 1.742 | 0.084 | -0.004 | 0.066 |
glance(discord_model) %>%
select(r.squared, adj.r.squared, sigma, p.value, nobs) %>%
knitr::kable(digits = 3)| r.squared | adj.r.squared | sigma | p.value | nobs |
|---|---|---|---|---|
| 0.102 | 0.069 | 0.978 | 0.012 | 141 |
stargazer::stargazer(discord_model,
discord_model_manual,
type = "html", ci = TRUE,
digits = 3, single.row = TRUE, title = "Discordant Regression Results Comparison"
)% Error: Unrecognized object type.
Both the manual and helper function approaches yield identical results.
Comparing the Three Approaches
To fully understand the value of discordant-kinship regression, it’s helpful to compare all three models side by side. Each approach answers a different question about the data.
Side-by-Side Model Comparison
stargazer::stargazer(
ols_model,
between_model,
discord_model_manual,
type = "html", ci = TRUE,
digits = 3,
single.row = TRUE,
title = "Comparison of OLS, Between-Family, and Discordant Regression Models",
column.labels = c("Standard OLS", "Between-Family", "Discordant"),
model.names = FALSE
)| Dependent variable: | |||
| weight | weight_mean | weight_diff | |
| Standard OLS | Between-Family | Discordant | |
| (1) | (2) | (3) | |
| height | 0.287*** (0.134, 0.440) | ||
| age | -0.073** (-0.129, -0.017) | ||
| weight_mean | -0.043 (-0.170, 0.084) | ||
| height_mean | 0.199** (0.049, 0.350) | 0.041 (-0.077, 0.158) | |
| height_diff | 0.141*** (0.056, 0.225) | ||
| age_mean | -0.044* (-0.089, 0.001) | 0.031* (-0.004, 0.066) | |
| age_diff | -0.026 (-0.074, 0.021) | ||
| Constant | 2.788*** (1.081, 4.495) | 2.064*** (0.759, 3.368) | 0.500 (-0.534, 1.534) |
| Observations | 141 | 141 | 141 |
| R2 | 0.125 | 0.067 | 0.102 |
| Adjusted R2 | 0.112 | 0.054 | 0.069 |
| Residual Std. Error | 1.573 (df = 138) | 1.284 (df = 138) | 0.978 (df = 135) |
| F Statistic | 9.857*** (df = 2; 138) | 4.970*** (df = 2; 138) | 3.060** (df = 5; 135) |
| Note: | p<0.1; p<0.05; p<0.01 | ||
Interpreting the Differences
For each model, we can summarize the key features, what they capture, and how to interpret the results:
Standard OLS Regression provides estimates of associations between variables at the individual level, controlling for measured covariates. However, it does not account for unmeasured familial confounding, which may bias the results. - Data: One sibling per family (e.g., first-born or older sibling)
Level of analysis: Individual level
What it captures: Total association between predictors and outcome, confounded by both genetic and environmental factors shared within families
Interpretation: “Individuals with higher height and age tend to have higher weight”
Between-Family Regression compares kin from different families, effectively controlling for all family-level unobserved heterogeneity. While it provides a clearer picture of the effects of interest, it may miss important individual-level variation within families. - Data: Average values for each kin pair
Level of analysis: Family level
What it captures: How family-average predictors relate to family-average outcomes
Interpretation: “Cousin pairs with higher average height and age tend to have higher average weight”
Discordant Regression focuses on within-family comparisons by examining kin with differing outcomes. It allows for a nuanced understanding of how individual-level factors interact above and beyond familial confounding. - Data: Differences between cousins within families
Level of analysis: Within-family (controlling for between-family variation)
What it captures: Within-family effects after controlling for shared genetic and environmental factors
-
Key coefficients:
*_mean: Between-family effects (similar to between-family model)*_diff: Within-family effects - the unique contribution of the predictor after controlling for familial confounding
Interpretation: “The cousin with higher weight tends to have higher height and age”
Advantage: Controls for unmeasured familial confounders, providing stronger evidence for (or against) causal effects # Conclusion
Discordant-kinship regression offers a powerful tool for disentangling within-family effects from between-family confounding. By leveraging sibling and cousin comparisons, researchers can gain insights into causal relationships that are often obscured in traditional analyses.
Session Info
sessioninfo::session_info()
#> ─ Session info ───────────────────────────────────────────────────────────────
#> setting value
#> version R version 4.5.2 (2025-10-31)
#> os Ubuntu 24.04.3 LTS
#> system x86_64, linux-gnu
#> ui X11
#> language en
#> collate C.UTF-8
#> ctype C.UTF-8
#> tz UTC
#> date 2026-02-21
#> pandoc 3.1.11 @ /opt/hostedtoolcache/pandoc/3.1.11/x64/ (via rmarkdown)
#> quarto NA
#>
#> ─ Packages ───────────────────────────────────────────────────────────────────
#> package * version date (UTC) lib source
#> backports 1.5.0 2024-05-23 [1] RSPM
#> BGmisc * 1.5.2 2026-01-11 [1] RSPM
#> broom * 1.0.12 2026-01-27 [1] RSPM
#> bslib 0.10.0 2026-01-26 [1] RSPM
#> cachem 1.1.0 2024-05-16 [1] RSPM
#> cli 3.6.5 2025-04-23 [1] RSPM
#> data.table 1.18.2.1 2026-01-27 [1] RSPM
#> desc 1.4.3 2023-12-10 [1] RSPM
#> digest 0.6.39 2025-11-19 [1] RSPM
#> discord * 1.3 2026-02-21 [1] local
#> dplyr * 1.2.0 2026-02-03 [1] RSPM
#> evaluate 1.0.5 2025-08-27 [1] RSPM
#> farver 2.1.2 2024-05-13 [1] RSPM
#> fastmap 1.2.0 2024-05-15 [1] RSPM
#> fs 1.6.6 2025-04-12 [1] RSPM
#> generics 0.1.4 2025-05-09 [1] RSPM
#> ggpedigree * 1.1.0.3 2026-01-11 [1] RSPM
#> ggplot2 * 4.0.2 2026-02-03 [1] RSPM
#> glue 1.8.0 2024-09-30 [1] RSPM
#> gtable 0.3.6 2024-10-25 [1] RSPM
#> htmltools 0.5.9 2025-12-04 [1] RSPM
#> htmlwidgets 1.6.4 2023-12-06 [1] RSPM
#> janitor * 2.2.1 2024-12-22 [1] RSPM
#> jquerylib 0.1.4 2021-04-26 [1] RSPM
#> jsonlite 2.0.0 2025-03-27 [1] RSPM
#> knitr 1.51 2025-12-20 [1] RSPM
#> labeling 0.4.3 2023-08-29 [1] RSPM
#> lattice 0.22-7 2025-04-02 [3] CRAN (R 4.5.2)
#> lifecycle 1.0.5 2026-01-08 [1] RSPM
#> lubridate 1.9.5 2026-02-04 [1] RSPM
#> magrittr * 2.0.4 2025-09-12 [1] RSPM
#> Matrix 1.7-4 2025-08-28 [3] CRAN (R 4.5.2)
#> NlsyLinks * 2.2.3 2025-08-31 [1] RSPM
#> otel 0.2.0 2025-08-29 [1] RSPM
#> pillar 1.11.1 2025-09-17 [1] RSPM
#> pkgconfig 2.0.3 2019-09-22 [1] RSPM
#> pkgdown 2.2.0 2025-11-06 [1] any (@2.2.0)
#> purrr 1.2.1 2026-01-09 [1] RSPM
#> quadprog 1.5-8 2019-11-20 [1] RSPM
#> R6 2.6.1 2025-02-15 [1] RSPM
#> ragg 1.5.0 2025-09-02 [1] RSPM
#> RColorBrewer 1.1-3 2022-04-03 [1] RSPM
#> rlang 1.1.7 2026-01-09 [1] RSPM
#> rmarkdown 2.30 2025-09-28 [1] RSPM
#> S7 0.2.1 2025-11-14 [1] RSPM
#> sass 0.4.10 2025-04-11 [1] RSPM
#> scales 1.4.0 2025-04-24 [1] RSPM
#> sessioninfo 1.2.3 2025-02-05 [1] RSPM
#> snakecase 0.11.1 2023-08-27 [1] RSPM
#> stargazer 5.2.3 2022-03-04 [1] RSPM
#> stringi 1.8.7 2025-03-27 [1] RSPM
#> stringr 1.6.0 2025-11-04 [1] RSPM
#> systemfonts 1.3.1 2025-10-01 [1] RSPM
#> textshaping 1.0.4 2025-10-10 [1] RSPM
#> tibble 3.3.1 2026-01-11 [1] RSPM
#> tidyr * 1.3.2 2025-12-19 [1] RSPM
#> tidyselect 1.2.1 2024-03-11 [1] RSPM
#> timechange 0.4.0 2026-01-29 [1] RSPM
#> vctrs 0.7.1 2026-01-23 [1] RSPM
#> withr 3.0.2 2024-10-28 [1] RSPM
#> xfun 0.56 2026-01-18 [1] RSPM
#> yaml 2.3.12 2025-12-10 [1] RSPM
#>
#> [1] /home/runner/work/_temp/Library
#> [2] /opt/R/4.5.2/lib/R/site-library
#> [3] /opt/R/4.5.2/lib/R/library
#> * ── Packages attached to the search path.
#>
#> ──────────────────────────────────────────────────────────────────────────────